CFTR variations and classification
According to the international recommendations, we should use the term "sequence variant" to indicate any sequence change, pathogenic or not. However, in medical genetics, the term "mutation" is widely used to indicate a "disease-causing sequence change". The term "polymorphism" indicates either a "neutral variant" (non disease-causing change) or a "change found in the general population at a frequency of ≥1%". These terms are imperfect since some "mutation" can result in mild or incomplete phenotypes or have an incomplete penetrance (asymptomatic individuals), and some "polymorphisms" (i.e. common CFTR variants) may contribute to the final amount of functional protein and determine the variable expressivity of complex alleles or haplotypes.
The Human Genome Variation Society (HGVS) proposes to use the term "affects or does not affect function" and to classify variants in 5 categories:
The Human Genome Variation Society (HGVS) proposes to use the term "affects or does not affect function" and to classify variants in 5 categories:
| Affects function |
|---|
| Probably affects function |
| Unknown: VUS or VUCS [variants of unknown (clinical) significance] |
| Probably does not affect function |
| Does not affect function |
CFTR-France uses the nomenclature recommended by HGVS (Human Genome Variation Society, http://www.hgvs.org/), notably:
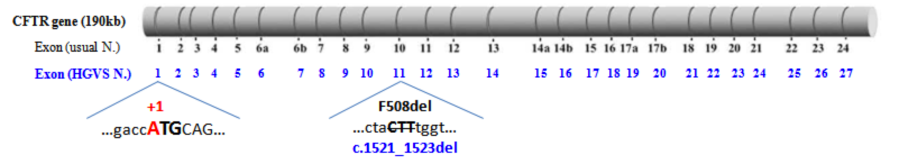
As an example, the disease-causing variation p.Phe508del corresponds to the nucleotide change c.1521_1523del and is located within exon 11 [instead of exon 10 in the historical exon numbering].
(Modified from Claustres et al., 2005)
- the 27 exons are numbered from 1 to 27 [instead of 1 to 24 in the historical numbering, with the three additional exons that were missed in the original publication of the gene designated as 6b, 14b, and 17b]
- the "A" of the translation initiation codon (ATG) corresponds to the position "+1" [instead of +133 in the usual nucleotide numbering used in the legacy nomenclature of variants].
As an example, the disease-causing variation p.Phe508del corresponds to the nucleotide change c.1521_1523del and is located within exon 11 [instead of exon 10 in the historical exon numbering].
Reference sequences
| CFTR gene | OMIM #602421 |
| Ensembl Gene ID | ENSG00000001626 |
| Chromosomal sequence | NC_000007.14 (excepted for position g.117559479: A is the wild type nucleotide, G a variant nucleotide) |
| Genomic sequence | NG_016465.3 (excepted for position g.98696: A is the wild type nucleotide, G a variant nucleotide) |
| Coding sequence | NM_000492.4 (excepted for position c.1408: A is the wild type nucleotide, G a variant nucleotide) |
| Protein sequence | NP_000483 (excepted for position 470: Methionine is the wild type amino acid, Valine is a variant amino acid) |
Download
- Schematic representation of the 27 CFTR exons with usual and HGVS numbering of nucleotides bordering exons. Amino acids phasing and corresponding domains of the CFTR protein (NBDs, R domain, ...) are also illustrated (pdf file).
At least 5 different types of alterations can be distinguished, based on the classification suggested by Castellani et al., 2008:
Go to next section “Classification of CFTR variants” to see the new CFTR-France classification.
Up to date, about 2,000 CFTR variations have been reported to the international CF database CFMD (Cystic Fibrosis Mutation Database). However, worldwide, less than 20 variants are found with a frequency ≥0.1% in the CF population (Castellani et al., 2008). The vast majority of CFTR disease-causing variations are rare or private, emphasizing the high allelic heterogeneity of the CFTR gene.
|
Named "disease-causing" in CFTR-France |
|
"Non disease-causing" in CFTR-France |
| "Unclassified variant" (VUS) in CFTR-France |
Frequent disease-causing variations
The most common disease-causing variation in populations from European descent is a deletion of 3 nucleotides, p.Phe508del (c.1521_1523del) (legacy name F508del). In France, this variant globally corresponds to 67% of alleles identified in patients with CF on average, with a wide regional variation: up to 50% of CF alleles in the most southern regions and almost 80% in Brittany (Claustres et al., 2000, Audrézet et al., 2014). The loss of only one amino acid (Phenylalanine at position 508) causes a severe misfolding of the CFTR protein. The mutant chloride channel shows an impaired glycosylation with a dramatic decrease in exocytic trafficking and peripheral stability, and is finally subjected to rapid proteasome-mediated degradation (p.Phe508del CFTR processing). In France, 3 other disease-causing variations have a frequency over 1%: p.Gly542* (G542X), c.1585-1G>A (1717-1G>A) and p.Asn1303Lys (N1303K). The frequency and type of mutants vary widely according to geographic and ethnic origin of individuals.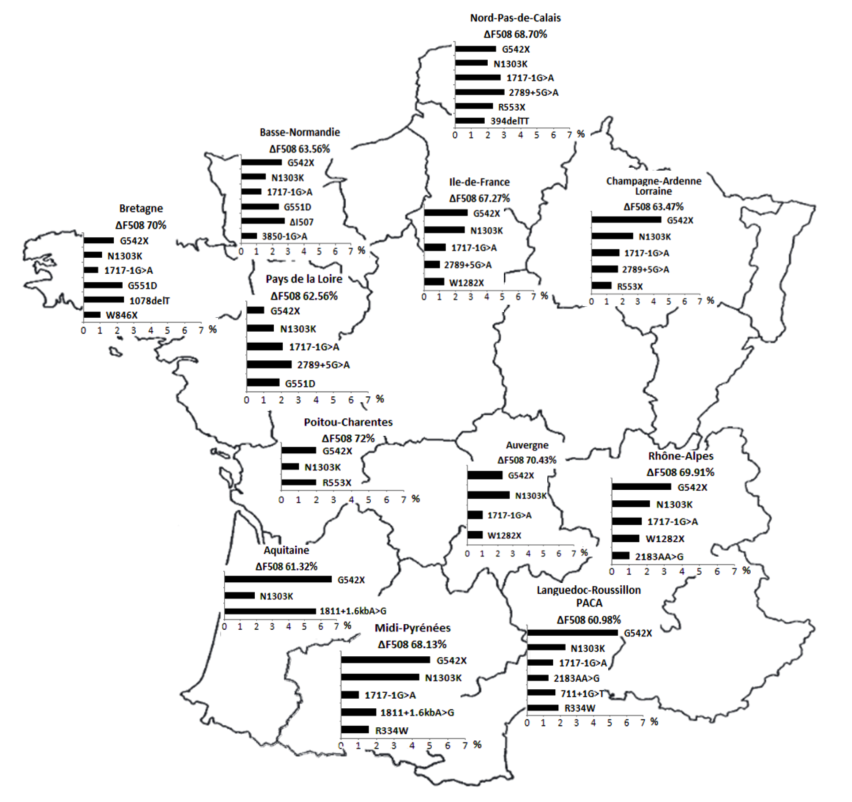
Rare variants
Only a few hundred of the 2,000 CFTR variants have been experimentally investigated and the disease liability of 159 most common variants found in the CF populations worldwide has been recently assessed for clinical severity, functional consequences and disease penetrance (Sosnay et al., 2013). This gap in the knowledge of clinical relevance of the vast majority of rare CFTR variants raises major issues in diagnosis and genetic counseling.
Classifications of CFTR variations based on experimental functional consequences or on computational methods that predict the impact of variations both help to interpret variants and to characterize their disease liability.
|
|

Remarks on the classification based on the functional impact of variations
1/ This classification proposed more than 20 years ago by Welsh & Smith (1993) was based on functional criteria related to chloride transport. It does not take into consideration the numerous other regulative roles of CFTR. 2/ Only a very limited number of mutations have been assessed for CFTR dysfunction in experimental assays and epidemiological studies, so some variants may be misclassified. 3/ Many CFTR variants have been shown to impair more than a single process; an example is the most common mutation, p.Phe508del, which also creates a gating defect (class III) amongst the few misfolded proteins that escape the proteasome degradation pathway and reach the apical membrane (Dalemans et al., 1991). 4/ Clinicians and pharmaceutical industry should consider updated information on the liability and classification of CFTR variants.A/ Classification based on the effect on CFTR gene or protein function
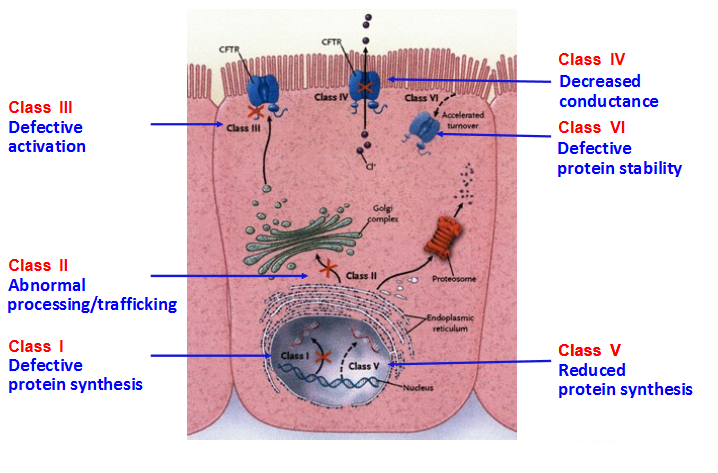
Class I: Defective protein synthesis, leading to absence of functional CFTR channel at the surface of epithelial cells. This class includes nonsense variants and variants that result in a premature termination codon (PTC) such as variations affecting canonical splice sites and insertions or deletions that cause a shift in the reading frame. PTC variants generally cause the degradation of mRNA by the control mechanism NMD (nonsense-mediated mRNA decay), leading to the absence of protein production; e.g: p.Arg553* (R553X), p.Gly542* (G542X), p.Trp1282* (W1282X). There are few exceptions where transcripts bearing a PTC are stable but the mutant protein is rapidly degraded; e.g: p.Arg1162* (R1162X). Class II: Defective processing and maturation, leading to absence of functional CFTR channel at the surface of epithelial cells. A number of mutations cause a defect in post-translational folding, maturation (incomplete glycosylation) and localization of CFTR, leading to its rapid degradation. Although there are ethnic and geographical variations, approximately 90% of European CF patients harbor a class II variant on at least one mutant allele, p.Phe508del accounting for the vast majority of them. Other class II variants are missenses such as p.Asn1303Lys (N1303K) or p.Arg560Thr (R560T). Class III: Defective channel regulation/gating, leading to a present but non-functional CFTR protein at the apical membrane. Some missense variations, mostly located in Nucleotide Binding Fold (NBD) domains, are properly processed and addressed to the membrane, but the amino acid substitution leads to markedly reduced anion conductance and resistance to activation (preventing channel opening). The most widely studied example is the "gating mutant" p.Gly551Asp (G551D). Class IV: Defective channel conductance, leading to a present CFTR protein at the apical membrane but with reduced anion conductance. Some missense variations, mostly located in trans-membrane domains involved in the formation of the channel pore, produce a CFTR protein with altered conductance properties such as ion selectivity. Examples include p.Arg347Pro (R347P), p.Arg117His (R117H). Class V: Mutations causing abnormal splicing, leading to a mix of CFTR mRNA correctly or aberrantly spliced, and thus to reduced amounts of normal CFTR protein. This class includes intronic variations such as "the 5T allele" (c.1210-12T[5]) or c.3718-2477C>T (3849+10kbC>T) and some missense variations altering protein trafficking (p.Ala455Glu) or located within the promoter. Class VI: Defective stability of CFTR protein, leading to absence or severe reduction of CFTR at the membrane surface. Some missense variants such as p.Asn287Tyr (N287Y) result in CF without affecting biosynthesis or channel gating: they accelerate endocytic retrieval from the plasma membrane and lead to a severe reduction in the steady-state level of CFTR at the cell surface (Silvis et al., 2003). Generally, classes I, II, III variants are considered as "severe" whereas classes IV, V and VI, which retain some residual CFTR function, are considered as "milder" variations. However, CFTR dysfunction can vary depending on organs, and may change over time.
B/ CFTR-France classification based on frequency in population, patients' data and functional data
The CFTR-France variants classification changed in 2024 and now includes five main classes, which can be transposed to the five ACMG classes, as demonstrated in the table below: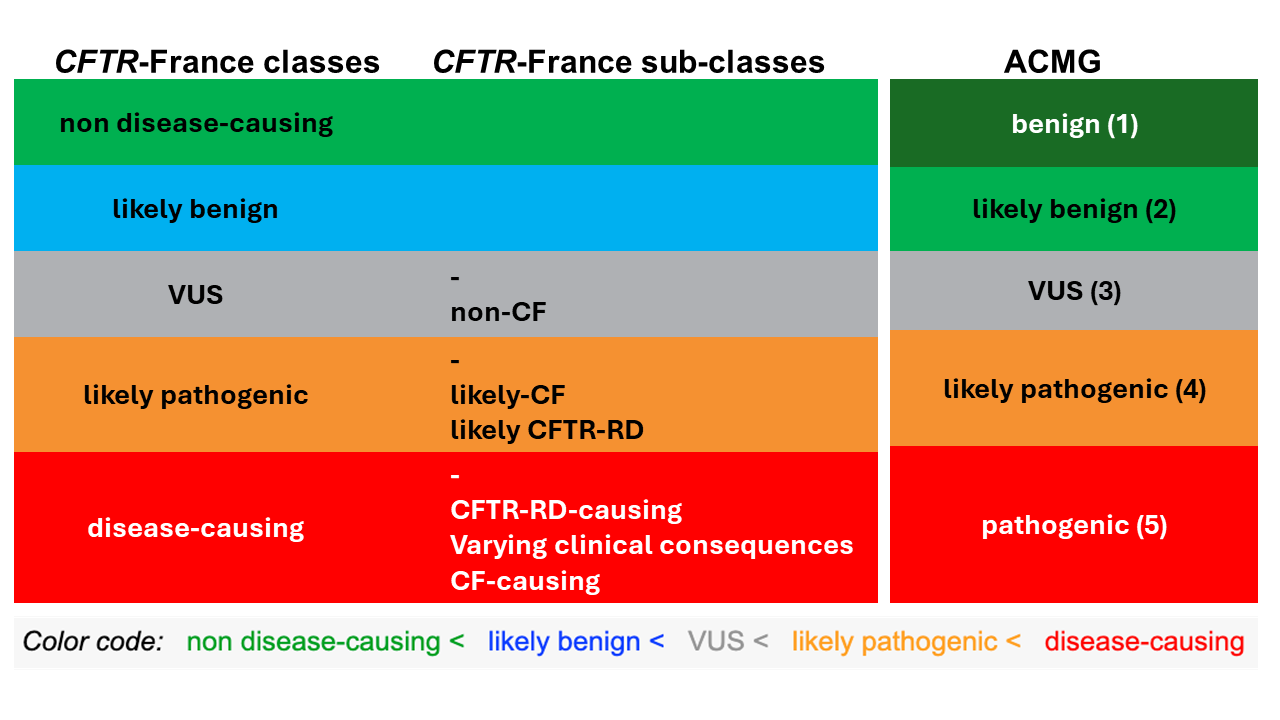
To address the issue of phenotype variability observed in patients, we completed this classification with subclasses that inform users about the associated phenotype based on the data available in CFTR-France, other databases, the literature, and in silico predictions (for VUS). - The “likely benign” class is based on epidemiological data (in particular high frequency in the general population), the number and type of diagnosis for patients reported in CFTR-France and/or functional data. As in most cases, the patients' data is not sufficient to definitely classify these variants as benign. - The “likely pathogenic” class includes two subclasses: “likely CF” or “likely CFTR-RD” depending on whether the variant has been identified in patients with cystic fibrosis or only in patients with CFTR-RD. If data are deemed insufficient for a variant, no subclass is be provided. - The “disease-causing” class has three subclasses: “CF-causing”, “CFTR-RD-causing” and “varying clinical consequence ”. If the variant is pathogenic but its phenotypic spectrum has not yet been determined (CF or CFTR-RD), no subclass is entered.Note: A specific “low penetrance“ annotation has also been added for certain variants identified in patients but also in asymptomatic individuals reported in CFTR-France, proving that the penetrance of a given phenotype is not always complete and sometimes low (few patients vs. asymptomatic individuals reported). It is the case for instance for the variant p.Leu997Phe (L997F) which is classified CFTR-RD with low penetrance. Similarly, the annotation “low penetrance for CF“ means that the variant is reported in one or a few patients with CF but that their number is very low compared to the CFTR-RD patients carrying the variant evaluated. For example, the variants p.Asp1152His (D1152H) or c.1210-34_1210-6TG[13]T[5] (TG13T5) are classified as varying clinical consequence with low penetrance of CF. - The “non-CF” subclass is therefore appended to the “VUS” class when only patients with CFTR-RD have been reported in CFTR-France. The available data are then sufficient to consider that the variant is very likely not associated with cystic fibrosis. This constitutes an important information for genetic counseling of relatives in particular for pregnancy management. As a reminder, the former CFTR-France classification was composed of 5 main classes based on the classification suggested by Castellani et al., 2008. Variants associated to classical forms of CF, variants associated to CFTR-RDs, variants with clinical varying consequences, neutral variants and unclassified variants (VUS). The VUS class used to have a 5-class system (ranging from VUS1 to VUS5). VUS1 was used to describe probably not pathogenic variants and VUS5 for probably pathogenic variants, as recommended by the Association for Clinical Genetic Science and the Dutch Society of Clinical Genetic Laboratory Specialists (2013). This classification was made by coupling different bioinformatics resources with functional splicing assays (hybrid minigenes) and epidemiological data, in order to predict the pathogenicity of CFTR variants of unknown significance (VUS) (Desmet et al., 2010; Sosnay et al., 2011; Raynal et al., 2013, Sosnay and Cutting, 2014).

